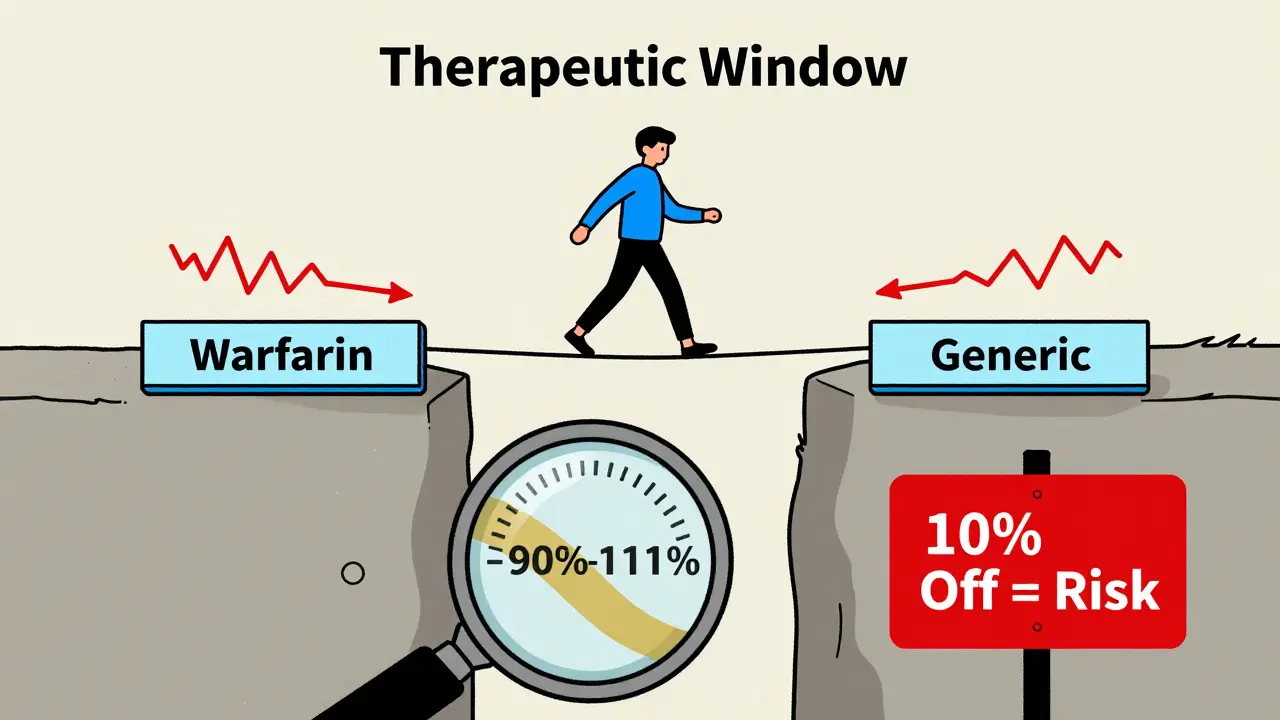
When a drug has a narrow therapeutic index (NTI), even tiny differences in how it’s absorbed by the body can mean the difference between healing and harm. Think of it like walking a tightrope - one step too far, and you fall. That’s why NTI generics aren’t just another copy of a brand-name drug. They demand a whole different level of scrutiny. Bridging studies are the backbone of that scrutiny. These aren’t your run-of-the-mill bioequivalence tests. They’re complex, expensive, and tightly controlled. And they’re absolutely necessary to make sure generic versions of drugs like warfarin, phenytoin, and levothyroxine are just as safe and effective as the originals.
What Makes NTI Drugs So Different?
Not all drugs are created equal when it comes to safety margins. Most medications have a wide buffer between the dose that works and the dose that causes harm. But NTI drugs? That buffer is razor-thin. The FDA defines an NTI drug as one where the difference between the minimum effective dose and the minimum toxic dose is no more than two-fold. That means if your dose is off by just 10%, you could be underdosed - and your condition flares up - or overdosed, and you could suffer serious side effects, even death.
Drugs like warfarin (a blood thinner), phenytoin (for seizures), digoxin (for heart failure), and levothyroxine (for thyroid conditions) fall into this category. They all require regular blood tests to monitor levels. Even small changes in how the body processes the drug can lead to dangerous swings. That’s why a generic version of warfarin can’t just be tested like a generic ibuprofen. The stakes are too high.
The Bridging Study Blueprint
For standard generics, regulators usually rely on a simple two-way crossover study: one group takes the brand drug, another takes the generic, and their blood levels are compared. But for NTI generics, that’s not enough. The FDA and other global agencies require a four-way, fully replicated crossover design. This means each participant takes both the brand and the generic drug twice - once in the morning, once in the evening - over several weeks. It’s exhausting for volunteers, expensive for manufacturers, and takes 12 to 18 months just to complete.
Why so complex? Because NTI drugs often show high variability in how they behave from person to person. A standard 80%-125% bioequivalence range - acceptable for most drugs - is too loose here. For NTI drugs, the acceptable range tightens to 90.00%-111.11% for both Cmax (peak concentration) and AUC (total exposure). That’s a much narrower window. And it’s not just about the numbers. The quality of the drug itself must be tighter. The active ingredient must be within 95%-105% of the labeled amount, compared to 90%-110% for non-NTI drugs.
Cost, Time, and Barriers to Entry
Developing an NTI generic isn’t just harder - it’s dramatically more expensive. A standard bioequivalence study might cost $1.5 million to $2.5 million. For NTI drugs? It’s $2.5 million to $3.5 million. Why? More subjects. Longer study duration. More blood draws. More data analysis. And that’s just the clinical part. The regulatory paperwork is 25%-30% thicker. Companies have to submit pharmacokinetic models, statistical justifications, and detailed dissolution profiles that meet exacting standards.
According to a 2022 survey by the Generic Pharmaceutical Association, 78% of manufacturers see NTI drug development as significantly more challenging than standard generics. Sixty-three percent pointed directly to bridging study requirements as the biggest roadblock. Teva Pharmaceuticals noted that a four-way crossover study requires twice as many volunteers and takes 40%-50% longer than a standard two-way study. That means more time, more money, and more risk of dropout - which can sink the entire trial.
And it shows in the numbers. Between 2018 and 2022, the FDA approved only 18 NTI generics. In the same period, it approved over 1,000 non-NTI generics. That’s not because there’s no demand. It’s because the path to approval is so steep. Thirty-seven percent of rejected NTI applications cited inadequate bridging study design as the main reason - compared to just 12% for non-NTI drugs.

Who’s in Charge? Regulatory Differences
The FDA leads the way with its strictest standards. But other agencies aren’t far behind. The European Medicines Agency (EMA) issued a 2022 position paper stating that NTI drugs require specific study designs - and those can’t be waived just because the product looks similar. The International Council for Harmonisation (ICH) has been working since the 1990s to align global standards, and by 2025, it aims to finalize a new guideline (E18) that addresses ethnic differences in how NTI drugs behave across populations.
Some industry groups, like the International Generic and Biosimilar Medicines Association (IGBA), have pushed for flexibility - suggesting that certain NTI drugs might be eligible for bridging study waivers under specific conditions. But regulators push back. Dr. Philip K. Robinson of the FDA’s Office of Generic Drugs put it plainly: “For NTI drugs, even minor differences in pharmacokinetics can have clinically significant consequences.”
Why Only 42% Market Share?
Despite the high demand for affordable versions of these life-saving drugs, generic penetration for NTI drugs sits at just 42%. Compare that to 85% for non-NTI generics. Why the gap? It’s not about patient preference. It’s about supply. Only a handful of manufacturers have the expertise, resources, and patience to tackle NTI development. The FDA estimates that only 35% of generic companies have in-house statisticians trained in reference-scaled average bioequivalence (RSABE) methods - the statistical engine behind these studies.
Building that expertise takes 18 to 24 months. Many smaller companies simply can’t afford the learning curve. And even when they do, the risk of rejection remains high. That’s why the FDA created its Complex Generic Drug Products Pilot Program - a streamlined review process for NTI and other hard-to-make generics. Participating companies saw review times drop by 25%.

The Future: Can Modeling Replace Clinical Trials?
One of the most exciting developments is the use of physiologically-based pharmacokinetic (PBPK) modeling. Instead of running full clinical studies, companies can simulate how a drug behaves in the body using computer models based on real-world data. In a 2022 FDA pilot study, PBPK modeling successfully predicted the behavior of warfarin generics without needing a full bridging trial. That’s promising.
But regulators aren’t ready to ditch clinical data yet. Dr. Sally Sepehrara of the FDA’s Office of Generic Drugs stated in 2023: “For the foreseeable future, robust clinical data will remain essential for NTI drug approval.” The models are getting better, but they still need validation through real human data. For now, bridging studies remain the gold standard.
What’s Next?
The global NTI drug market was worth $78.5 billion in 2022 and is expected to grow to over $100 billion by 2028. Yet, only a fraction of that is served by generics. That’s a $32.8 billion opportunity waiting to be unlocked - if manufacturers can overcome the barriers.
For patients, this means more affordable access to critical drugs like levothyroxine and warfarin. For regulators, it means balancing safety with access. For manufacturers, it means investing in specialized expertise - or partnering with those who already have it.
The bottom line? NTI generics aren’t just another product line. They’re a test of science, regulation, and commitment to patient safety. And until we find a way to make these studies faster, cheaper, and just as reliable, the gap between brand and generic will remain - not because we can’t make the drugs, but because we refuse to cut corners on safety.
Why are bridging studies required for NTI generics but not for all generics?
Bridging studies are required for NTI generics because these drugs have a very small window between the effective dose and the toxic dose. Even minor differences in how the body absorbs the drug - like a 5% change in concentration - can lead to serious health risks. Standard generics don’t need this level of scrutiny because their safety margins are wide. For NTI drugs, regulators use tighter bioequivalence criteria (90%-111%) and more complex study designs to ensure the generic performs almost identically to the brand.
What drugs are considered NTI drugs?
Common NTI drugs include warfarin, phenytoin, digoxin, levothyroxine, lithium, and cyclosporine. These drugs require precise dosing and often routine blood monitoring because their therapeutic and toxic levels are very close. The FDA has officially listed 27 specific products as NTI drugs in its 2023 draft guidance, and more may be added as research evolves.
How do bridging studies for NTI generics differ from standard bioequivalence studies?
Standard bioequivalence studies use a two-way crossover design with an 80%-125% acceptance range. NTI bridging studies use a four-way, fully replicated crossover design with a much tighter 90.00%-111.11% range. They also require more subjects, longer study durations, and specialized statistical methods like reference-scaled average bioequivalence (RSABE). The goal is to account for natural variability in how patients process these drugs while still ensuring safety.
Why are NTI generics so expensive to develop?
Developing an NTI generic costs 30%-50% more than a standard generic - typically $2.5 million to $3.5 million versus $1.5 million to $2.5 million. This is due to longer, more complex clinical trials (12-18 months vs. 6-9 months), higher subject recruitment needs, stricter quality controls, and more extensive regulatory documentation. Only a small number of manufacturers have the expertise to handle these studies, limiting competition and driving up costs.
Can PBPK modeling replace bridging studies for NTI generics?
PBPK modeling shows promise and has been successfully tested in FDA pilot studies with drugs like warfarin. It uses computer simulations to predict how a generic will behave in the body. However, regulators still require clinical data to validate these models. While modeling may reduce the need for full clinical trials in the future, the FDA states that robust clinical evidence will remain essential for NTI drug approval for the foreseeable future.
13 Comments

Byron Duvall
March 3 2026
they're just making this up to keep prices high. i bet the brand names pay off the fda to block generics. same old story.

Full Scale Webmaster
March 3 2026
Let me tell you something. This whole NTI thing is a scam. I’ve been reading up on this for weeks and I’m convinced the FDA is being manipulated by Big Pharma. Why do you think they’re so adamant about four-way crossover studies? Because it’s expensive. And expensive means fewer competitors. And fewer competitors means they can keep charging $500 for a 30-day supply of warfarin. It’s not about safety-it’s about profit. They’ll drag out studies for years, then claim ‘we need more data’ while the brand sits on monopoly pricing. And don’t even get me started on how they cherry-pick ‘bioequivalence’ metrics. Cmax? AUC? What about real-world outcomes? No one’s measuring hospitalizations. No one’s tracking deaths. They’re hiding behind statistics while patients suffer. This isn’t science. It’s a corporate shell game.

Angel Wolfe
March 4 2026
this is why america is falling apart. we let foreigners make our medicine. if we had our own labs and our own scientists we wouldn't need these stupid bridging studies. just make the drug here and be done with it. why do we trust india and china to make our life saving pills? they don't even have the same values as us. this is national security.

Sophia Rafiq
March 5 2026
RSABE is the real MVP here. Honestly, the stats behind NTI generics are wild. You’re not just comparing means-you’re modeling variability across populations, accounting for food effects, circadian rhythms, even gut microbiome shifts. It’s not just bioequivalence. It’s bio-precision. And yeah, it costs more. But when your drug is literally the difference between a seizure and a Sunday, you don’t cut corners.

Martin Halpin
March 5 2026
You know what’s funny? People act like this is some new breakthrough. Back in 2010, I worked on a phenytoin generic project. We had to run 18 months of studies. 120 subjects. Blood drawn every 30 minutes for 72 hours. One volunteer dropped out after day 5 because he said he felt ‘like a lab rat with a IV in his arm.’ And you know what? The final data showed a 107% Cmax difference. The FDA rejected it. So we tried again. And again. And again. Now? We have a product on the market. But it cost $4.2 million. And we’re still the only ones making it. This isn’t regulation. It’s a barrier to entry disguised as science.

Eimear Gilroy
March 7 2026
So if PBPK modeling works for warfarin, why not expand it? Is it really about safety-or about inertia in regulatory systems? I mean, we use AI to predict weather patterns with 95% accuracy. Why can’t we simulate drug absorption with the same rigor? Are we scared of innovation?

Ajay Krishna
March 8 2026
I come from a country where many people can't even afford basic meds. Seeing how strict the process is for NTI generics makes me hopeful. It means someone cares enough to protect people, even if it's expensive. I wish more countries adopted this. Not because it's hard-but because it's right. We don't need cheaper drugs. We need safe ones. And if that costs more, so be it. Lives > profits.

Charity Hanson
March 8 2026
This is why I love science. Real, messy, complicated, expensive science. No shortcuts. No magic bullets. Just people working hard to make sure someone’s mom doesn’t have a stroke because a pill was 8% off. Huge respect to the teams doing this. Keep going. The world needs more of this.

Sneha Mahapatra
March 8 2026
sometimes i think we forget that behind every dosage pill is a human who depends on it. not a statistic. not a market segment. a person. maybe we don't need to rush. maybe we need to slow down and get it right. 🌱

Miranda Anderson
March 10 2026
I’ve been in pharma for 18 years. I’ve seen generics get approved for everything from antihistamines to statins. But NTI? That’s a whole different beast. The one thing no one talks about? The quality control. The raw material. Even a 1% impurity in the active ingredient can throw off the whole profile. That’s why the 95-105% purity requirement exists. It’s not bureaucracy. It’s chemistry. And chemistry doesn’t lie.

Gigi Valdez
March 10 2026
The regulatory framework for NTI generics is not merely a technical requirement-it is a necessary safeguard grounded in pharmacological principles. The narrowing of bioequivalence thresholds reflects an evidence-based approach to minimizing clinical risk. While cost and time are legitimate concerns, they must be weighed against the potential for iatrogenic harm. Therefore, the current paradigm, though resource-intensive, remains scientifically defensible and ethically imperative.

bill cook
March 11 2026
you know what's worse than the fda? the fact that no one is talking about how these studies are run in eastern europe now. they're using cheap labor, rushed protocols, and data that's been 'adjusted.' you think your generic warfarin is safe? think again. they're cutting corners. and you're the one taking it.
Write a comment
Popular Posts



Aisling Maguire
March 1 2026
I work with NTI drugs daily and honestly? This post nailed it. That 90-111% range isn't just a number-it's the difference between someone going to the ER or staying home. I've seen cases where a 5% shift in levothyroxine levels caused full-blown hypothyroid symptoms. These studies suck for manufacturers, but they save lives. No compromises here.